[Extracellular Glycation and Crosslinks]
[Legendary Home Page] [Company Information] [Research Program]
[Background: The Biology of Aging]
[Scientific Advisory Board] [Contact Information]
Background: Introduction to
the Biology of Aging and Senescence
John D. Furber
The study of human senescence has been fraught with controversy, conflicting theories, and puzzling data. Gerontologists do not even agree on whether "pure" senescence is distinct from diseases of old age. Medical science has cataloged many signs of senescence. It manifests as dozens of changes in cells, tissues, and organs during aging. Human life is supported by a complex network of biochemical substances and reactions which affect the physical state and vitality of the body and mind. Senescent changes can be seen in the rate and outcome of many of these reactions. However, many of these changes are secondary effects of senescence, rather than primary causes. A summary of some of the secondary effects in human aging can be found in the Dossier on Ageing prepared by the Health on the Net Foundation.
Most of the biochemical reactions of life were developed long ago in single-celled organisms and bacteria, long before multicellular organisms arose. These single-celled organisms reproduced by dividing into two equal halves. Neither half was parent or child. Some of their descendants are still thriving today, living and dividing, and apparently not senile. Consequently, it is attractive to think of these protista and monera as never aging. However, there is evidence that the processes of growing and dividing are important factors in maintaining the youthful state of these cells.
When multicellular organisms develop, some of the cells (germ-line cells) are destined to become sperm or egg for the next generation. Other cells make up the body (soma cells), but will never become part of the next generation. In humans, some types of soma cells never divide again after reaching maturity (postmitotic or nonmitotic cells). The brain, skeletal muscles, and heart contain large numbers of postmitotic cells. Our single-celled ancestors never lived as postmitotic cells, so they never had to evolve mechanisms for postmitotic immortality. Why not? Well, if they stopped dividing, then they would not have progeny, so they could not be our ancestors.
Because multicellular organisms are able to reproduce with germ-line cells before senescence of their soma incapacitates them, there was never any evolutionary impetus to develop biochemical mechanisms of preventing senescence in their postmitotic cells. The germ-line lives on in the next generation while the bodies of the former generations senesce and die. This scenario is called the Disposable Soma Theory for the Evolution of Senescence. It is useful for understanding the ancient origins of senescence, but it does not exactly explain how the mechanisms of senescence work nor how to design interventions. Nevertheless, it does suggest taking a careful look at gradual changes in postmitotic tissues over the lifetime of the organism. Furthermore, it does not preclude the possibility that several primary processes could be acting in parallel to produce senescence effects.
In the following discussion, the interactions among these mechanisms may be better kept in mind by referring to the chart below. [ Click on it for a larger view. ]Disposable Soma Theory for the Evolution of Senescence
It is noteworthy that the germ line of egg and sperm has been maintained alive and safe from senescence and oxidative decay for over a billion years. Our life is part of an unbroken chain of life, extending back in time to our earliest ancestors. -- J.D. FurberMechanisms of Senescence
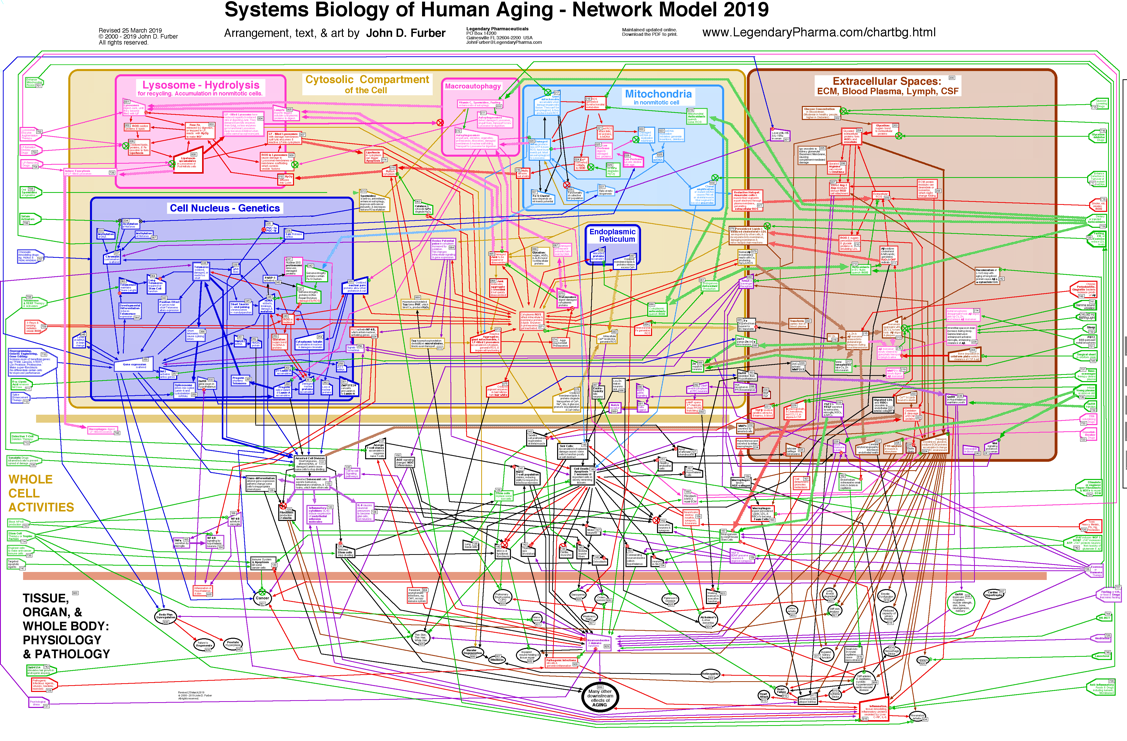
[ Click here for a larger view. ]
In defense, cells produce endogenous enzymes which prevent and repair some of this oxidative damage. Among these are superoxide dismutase and catalase, which detoxify some of the reactive chemicals. Dietary antioxidants, such as vitamin C, vitamin E, N-acetyl-cysteine, and alpha-Lipoic Acid, might also be important protective factors. However, many dietary supplement do not ever travel to the subcellular mitochondria and lysosomes where most free radicals exist. Furthermore, homeostatic mechanisms may downregulate endogenous antioxidants whenever exogenous antioxidants quench some of the free radicals. In cell culture experiments, antioxidants can protect cells somewhat from oxidizing chemicals and radiation. However, so far, observed benefits to humans taking large doses of dietary antioxidants have been limited. Wallace has presented the hypothesis that these diseases and many symptoms of senescence are the consequences of dysfunctional mitochondria, which contain mutations in their mtDNA, which reduce the efficiency of ATP production, and which result in the production of excessive concentrations of reactive chemicals. These ROS cause further damage to mitochondria and mtDNA, resulting in a positive feedback loop which worsens the situation. In some cases, the stressed mitochondria initiate a programmed cell death. In other cases, they signal the nucleus to divide and may initiate cancer. Meanwhile, some of the ROS leak out of the mitochondria and damage other cellular components.
De Grey presents an alternative hypothesis (the Reductive Hotspot Hypothesis) which places more emphasis on export of reactive chemicals from a few cells which have lost all oxidative phosphorylation function through clonal amplification of mutant mtDNA within those cells.
Further information may be found in:
Lipofuscin may also cause lysosomes to leak degradative enzymes and ROS into the cytoplasm, causing further damage to cellular components.
Further information may be found in:
Glycation and crosslinking contribute strongly to many progressive diseases of aging, including atherosclerosis, cardiovascular diseases, kidney disease, stiffness of joints and skin, cataracts, impaired wound healing, complications of diabetes, and Alzheimer's Dementia.
[ For more detail about Glycation and Crosslinking <<< Click here. ]
Furber JD. "Repairing Extracellular Aging and Glycation,"
Chapter 19 in
Because these reactions accumulate damage more slowly than glycation, it is appropriate to focus therapy development efforts first on glycation and crosslinking.
Lifestyle and Environment
Proper nutrition and exercise can have profoundly beneficial effects upon human health. However, while unhealthy lifestyle and environmental stresses can contribute to senescence and other diseases, they are not solely responsible for senescence. Note that regardless of how virtuous their lifestyle or how pristine their environment, we have no reliable contemporary reports of humans living past 123 years [Austad, Walford]. On the other hand, animal research has shown significant lifespan extension and improved health in old age can be attained through calorie restriction, provided that good levels of micronutrients, protein, and antioxidants are maintained. The reasons are still under study, but may be related to lower blood sugar levels which reduce glycation, altered mitochondrial function which reduces production of free-radicals, or altered insulin receptor signaling.Oxidative Stress and Free-radicals (ROS)
There is strong evidence for the hypothesis that reactive chemicals and radiation can impair health and shorten lifespan through oxidative modifications and cross-linking which eventually drags the physiological machinery to a halt [Lin, de Grey]. Several species of highly reactive chemicals, called free radicals or reactive oxygen species (ROS) are by-products of normal metabolism. They include superoxide, hydrogen peroxide, and perhydroxyl radical. They are produced primarily in the mitochondria and the lysosomes.Redox Potential Poise
Another consequence of increased ROS concentration in a cell, or increased numbers of oxidatively modified lipids and proteins, is that the chemical equilibrium or poise of several redox couples is altered. This alters signaling pathways and enzyme activities, which in turn can alter chromatin conformation, alter gene expression, change the cell's differentiation state, send signals to other cells, or even intiate cell death.
Mitochondrial Deterioration
Several diseases associated with senescence appear to be the direct result of cells containing dysfunctional mitochondria. The typical human cell has several hundred mitochondria. Mitochondria are the sites of three important processes which are involved in the progression of senescence:
Mitochondria function to convert the chemical energy from food into ATP, which cellular enzymes use to do work. As a by-product of energy conversion, ROS are produced. Mitochondria are also involved in sensing when a cell is sick enough to initiate programmed cell death (apoptosis or necrosis). Mutated mitochondrial DNA (mtDNA) can impair energy conversion and increase ROS production. Increased amounts of mutated mtDNA have been associated with progeriatric syndromes (premature symptoms of aging), Alzheimer's disease, diabetes, deafness, heart failure, optic nerve degeneration, cancer, and several progressive muscle diseases, as well as with the aging process itself. [Wallace, Lodish, Stryer, de Grey, Fliss] Conversely, less than usual accumulated mtDNA damage has been found in animals who live longer and healthier as a result of calorie restricted diets.
Lysosomes and Lipofuscin
The recycling centers inside of cells are lysosomes. Tiny bags of digestive enzymes sealed in a membrane skin, these organelles take apart damaged or unneeded proteins, lipids, and mitochondria. However, occasionally, the lysosome will take in an abnormal chemical structure which resists degradation by lysosomal enzymes. This just stays inside the lysosome, eventually joined by other abnormal structures. Iron atoms in the lysosome produce streams of ROS, which chemically crosslink the abnormal structures together into larger masses. During the lifespan of an animal, lysosomes in postmitotic cells accumulate a lot of this highly cross-linked organic substance called "lipofuscin" or "ceroid". Late in life, many of these lysosomes have so much lipofuscin that they are less able to recycle the proteins, lipids, and mitochondria. Consequently, damaged mitochondria accumulate in these cells, lowering ATP production and increasing ROS production. Furthermore, oxidatively damaged enzymes accumulate in the cytosol, reducing the rate of essential cellular functions.
Aging of the Extracellular Matrix
Each cell is surrounded by other cells, and by connective fibers, and other materials referred to as the extracellular matrix or ECM. The matrix and surrounding cells form a living space, or niche for the cell. Over time, the connective fibers are seen to slowly and gradually break down and stick together. A major factor in this deterioration is the chemical process of glycation.
Glycation and Cross-Linking
Sugar molecules in the blood and in the cells chemically bond to proteins and to DNA. (This bonding is called "glycation", "the Maillard reaction", "the browning reaction", or "nonenzymatic glycosylation"). The process happens gradually, so that glycation accumulates over the years on the longest-lived proteins which do not get recycled very often. The clearest evidence of this is found in the extracellular collagen and elastin. Over time, in the presence of reactive oxygen species (ROS), glycation promotes covalent crosslinking between adjacent protein strands. Crosslinking reduces the flexibility, elasticity, and functionality of the proteins. Furthermore, the chemical modifications of glycation and crosslinking can initiate harmful inflammatory and autoimmune responses. Non-glycation deterioration of the ECM
Besides glycation, several other chemical reactions slowly degrade the ECM. These processes are outlined in the figures below. A detailed review of all of these processes, including glycation and crosslinking, is found in Chapter 19 of The Future of Aging.
The Future of Aging: Pathways to Human Life Extension
Ed. by G.M. Fahy, et.al.(Springer Science+Business Media B.V. 2010) DOI 10.1007/978-90-481-3999-6_19.
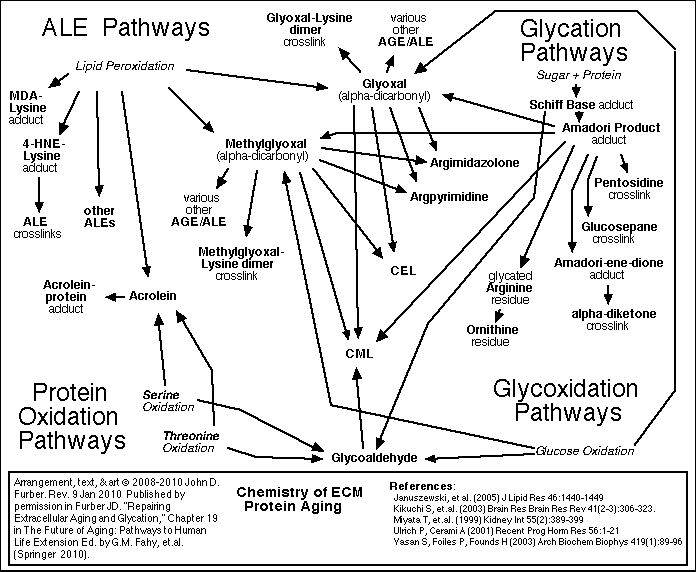
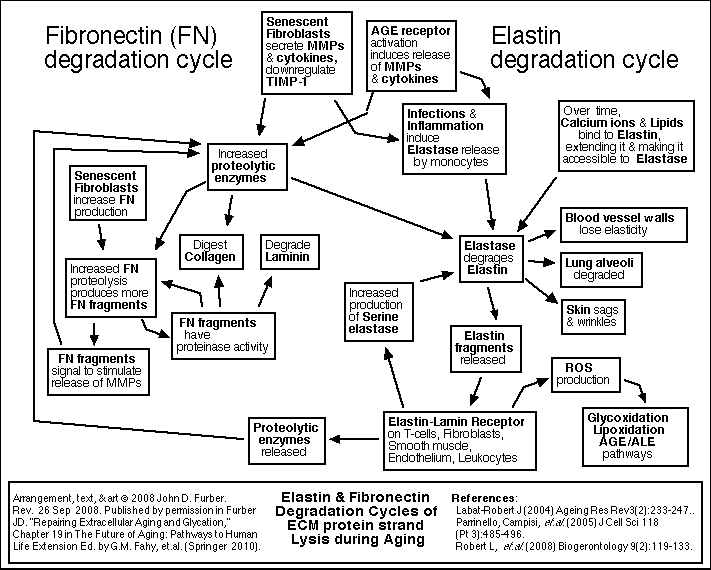
Cellular Senescence, Telomeres, and Telomerase
The telomerase gene is turned off in many adult human cells. As a result, these cells lose a bit off the ends (telomeres) of their chromosomes each time they divide. This appears to be related to their finite replicative lifespan in cell culture (The Hayflick Limit). Researchers are seeking to determine the importance of this observation to human senescence and health. One hypothesis suggests that as telomeres in some cells become very short, normally suppressed genes would be activated (see "Chromatin changes"), causing these cells to become "senescent". As a result, this small population of senescent cells would stop dividing and might export harmful signaling molecules (perhaps interleukins) to other cells in the body, resulting in some of the deleterious effects of human aging. In some cases, cells might "dysdifferentiate" or change to a kind of cell inappropriate for their position. Nevertheless, mice have longer telomeres than humans, but shorter lifespans, so aging in mice would seem to have other causes.Further information may be found in:
- K. Hopkin. "More Than a Sum of Our Cells," (3 October 2001) SAGE KE
Insulin receptor signaling
Side-effects of intracellular molecular signals from the insulin receptors affect the lifespan of the nematode worm, C. elegans. It is hypothesized that they may affect human health and lifespan as well. It has been suggested that lower insulin levels in calorie restricted animals might be partly responsible for their improved health and longer lives. This remains to be proven, as calorie restricted animals also have lower blood sugar levels, less glycation, and perhaps reduced ROS production.Further information may be found in:
- L. Guarente and C. Kenyon. "Genetic pathways that regulate aging in model organisms," Nature. Vol. 408 (9 Nov 2000) pp. 255-262.
Chromatin changes and gene expression
Changes in chromatin structure can alter access to regions of DNA by enzymes and ROS. Consequently, the expression of certain genes, and the probability of DNA damage and repair in these regions are altered when the chromatin structure changes. Chromatin changes are mediated by histone acetylation and DNA methylation, which are a cause and a result of gene expression and differentiation. It has been hypothesized that some of these changes might accumulate with age, perhaps in a small population of senescent cells which may spread damage throughout the body by malfunctioning or dying or by secreting damaging chemicals. Chromatin changes may also make the nuclear DNA more accessible to mutation events or to activation of cancer-promoting genes.Further information may be found in:
- J. Vijg.
Aging of the Genome: The Dual Role of DNA in Life and Death
(Oxford University Press, 2007).
- L. Guarente and C. Kenyon. "Genetic pathways that regulate aging in model organisms," Nature. Vol. 408 (9 Nov 2000) pp. 255-262.
- H Lodish, A Berk, SL Zipursky, P Matsudaira, D Baltimore, J Darnell.
Molecular Cell Biology
(W.H. Freeman & Co., Fourth Ed., 2000). pp.320-324, 384-389.
Stem cell stasis
If all stem cells were prevented from dividing, then tissue turnover and replacement of dying cells would be prevented. This could result in declines in functionality of many vital systems in the body. It has been hypothesized that stem cell division might be halted by either telomere shortening, chromatin modification, nuclear mutation, or altered hormonal signaling. This field is in need of further study. Stem cell stasis probably contributes to the deterioration of human aging. Progress in stem cell therapy could improve wound healing, tissue repair, and the treatment of many diseases. Further information may be found in:- K. Hopkin. "More Than a Sum of Our Cells," (3 October 2001) SAGE KE
Reading and Reference
Review Article
-
Jan Vijg & Judith Campisi. (2008)
Puzzles, promises and a cure for ageing.
Nature, 454, 1065-1071.
Video Documentaries
- Never Say Die is a Scientific American Frontiers documentary (Show # 1003) which first aired on January 25, 2000 on PBS television. [ Click here for Online Transcript at PBS. ]
- Stealing Time - The New Science Of Aging [VHS]
is a three-hour PBS documentary released in June 1999. The videotapes are no longer available from PBS Home Video, but can be found on Amazon.com
Books
- Steven N. Austad.
Why We Age: What Science Is Discovering about the Body's Journey Through Life
(John Wiley & Sons, 1997).
- Caleb E. Finch.
The Biology of Human Longevity:: Inflammation, Nutrition, and Aging in the Evolution of Lifespans
(Academic Press, 2007).
- Caleb E. Finch.
Longevity, Senescence, and the Genome
(University of Chicago Press, 1990).
- Enrique Cadenas and Lester Packer (Editors). Understanding the Process of Aging. Oxidative Stress and Disease series. (Marcel Dekker, Inc., 1999).
- J. Vijg.
Aging of the Genome: The Dual Role of DNA in Life and Death
- Aubrey de Grey. The Mitochondrial Free Radical Theory of Aging. (R.G. Landes Co., 1999).
- H Lodish, A Berk, SL Zipursky, P Matsudaira, D Baltimore, J Darnell. Molecular Cell Biology (W.H. Freeman & Co., Fourth Ed., 2000).
- Lubert Stryer. Biochemistry (W. H. Freeman & Co., Fourth Ed., 1995) p. 537.
- Roy L. Walford. Maximum Life Span (WW Norton & Co., 1983).

Hot Topics Navigation
[Network Flow Chart - Systems Biology of Aging]
[Extracellular Glycation and Crosslinks]
[Research Program] [Scientific Advisory Board] [Contact Information]
We are a participant in the Amazon Services LLC Associates Program, a program designed to provide for sites to earn fees by linking to amazon.com. As an Amazon Associate, we earn from qualifying purchases.
Rev. 16 January 2024 © 2000 - 2026 by John D. Furber. All rights Reserved.